


هموگلوبینوپاتی
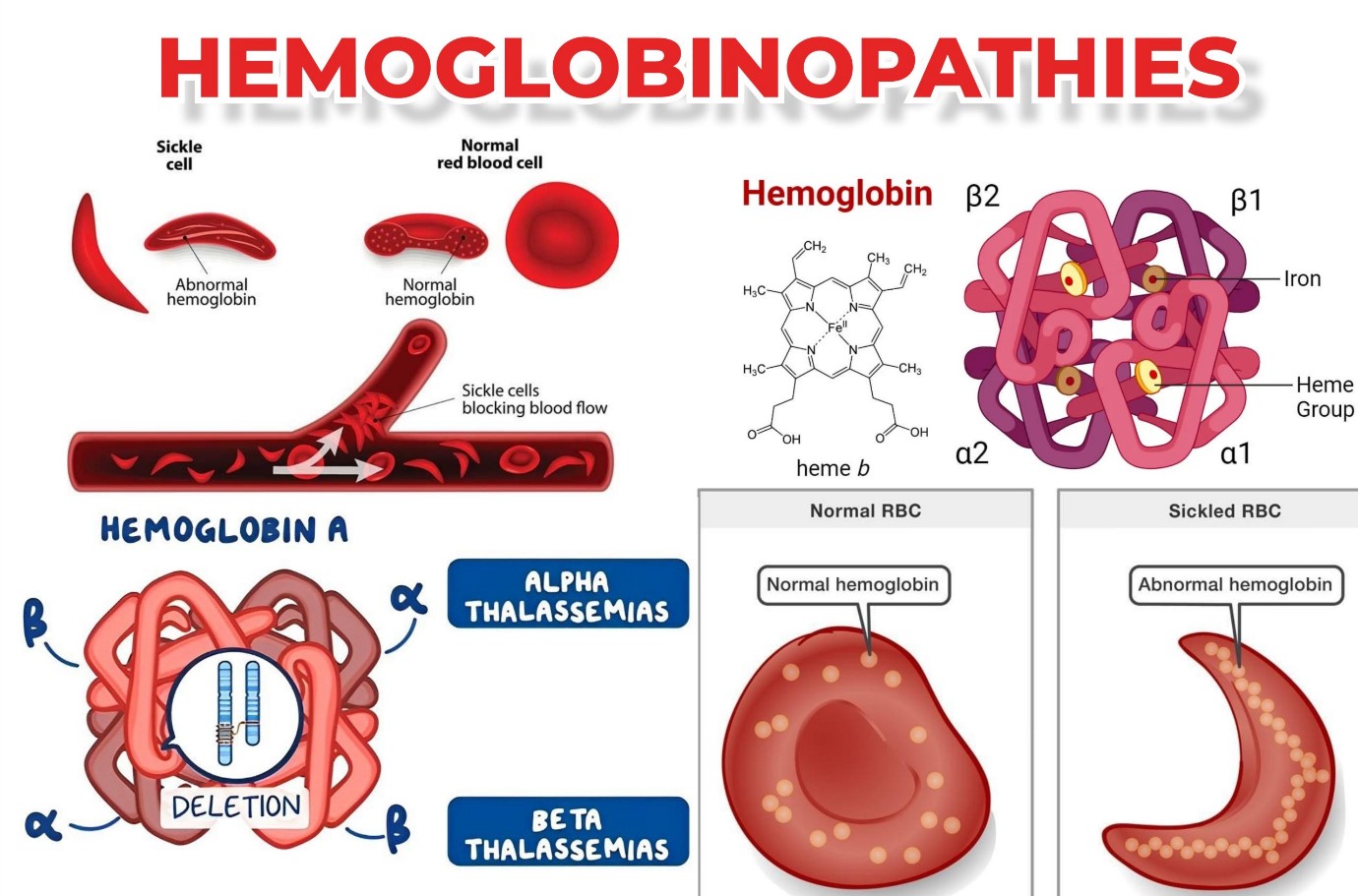
هموگلوبینوپاتی ها اختلالات ژنتیکی هستند که بر ساختار یا تولید هموگلوبین تأثیر می گذارند.
هموگلوبین (Hb) پروتئین حاوی آهن است که در تمام گلبولهای قرمز خون (RBCs) یافت میشود و با اتصال به مولکول اکسیژن در ریهها به گلبولهای قرمز اجازه میدهد تا اکسیژن را به تمامی سلولها و بافتهای بدن برساند. هموگلوبین شامل یک بخش به نام هِم، مولکولی با آهن مرکزی و بخش دیگری متشکل از چهار زنجیره گلوبین (پروتئین) است. زنجیره های گلوبین بسته به ساختارشان دارای نام های مختلفی هستند: آلفا، بتا، گاما و دلتا. نوع زنجیره های گلوبین در عملکرد هموگلوبین و توانایی آن در انتقال اکسیژن نقش اساسی دارند.
انواع طبیعی هموگلوبین عبارتند از:
- هموگلوبین A : حدود 95 تا 98 درصد هموگلوبین موجود در بزرگسالان را تشکیل می دهد که شامل دو زنجیره ی آلفا و دو زنجیره ی بتا است.
- هموگلوبین : A2 حدود 2 تا 3 درصد هموگلوبین را در بزرگسالان تشکیل می دهد. دارای دو زنجیره پروتئین آلفا و دو زنجیره دلتا است.
- هموگلوبین F هموگلوبین جنینی: 1تا 2 درصد هموگلوبین موجود در بزرگسالان را تشکیل می دهد. دارای دو زنجیره ی آلفا و دو زنجیره ی گاما است. این هموگلوبین اولین هموگلوبین تولید شده در دوران بارداری است. تولید آن معمولاً اندکی پس از تولد کاهش می یابد و در 1-2 سالگی به سطح بزرگسالان می رسد.
اصطلاح "هموگلوبینوپاتی" شامل تمام اختلالات ژنتیکی هموگلوبین است که به دو گروه اصلی به شرح زیر تقسیم می شوند که هر دو اختلال در اثر جهش یا حذف و یا ترکیبی از این دو در ژن α یا β گلوبین ایجاد می شوند:
سندرم های تالاسمی: هنگامی ست که نقص ژن باعث اختلالات سنتز Hb می شود، این امر منجر به تالاسمی می شود. ساختار هموگلوبین در این موارد طبیعی است.
- اختلالات ساختاری هموگلوبین (هموگلوبین های غیر طبیعی). هنگامی ست که تغییرات در ساختار Hb منجر به ایجاد هموگلوبین غیر طبیعی می شود .
- همچنین بسیاری از اشکال ترکیبی وجود دارد که ترکیبی از ویژگیهای هر دو گروه است، به عنوان مثال ترکیب تالاسمی آلفا- هموگلوبین E
سندرم های تالاسمی
شامل اختلالات سنتز Hb است .سندرم های تالاسمی ناشی از تولید ناکافی 1 یا 2 نوع زنجیره گلوبین است و با نوع آن (آلفا، بتا، دلتا، گاما) و میزان تولید ناکافی هموگلوبین (تعداد ژن های معیوب) و شدت علائم بالینی (جزئی، میانی، عمده)مشخص می شود. .
انواع سندرمهای تالاسمی :
- آلفا تالاسمی
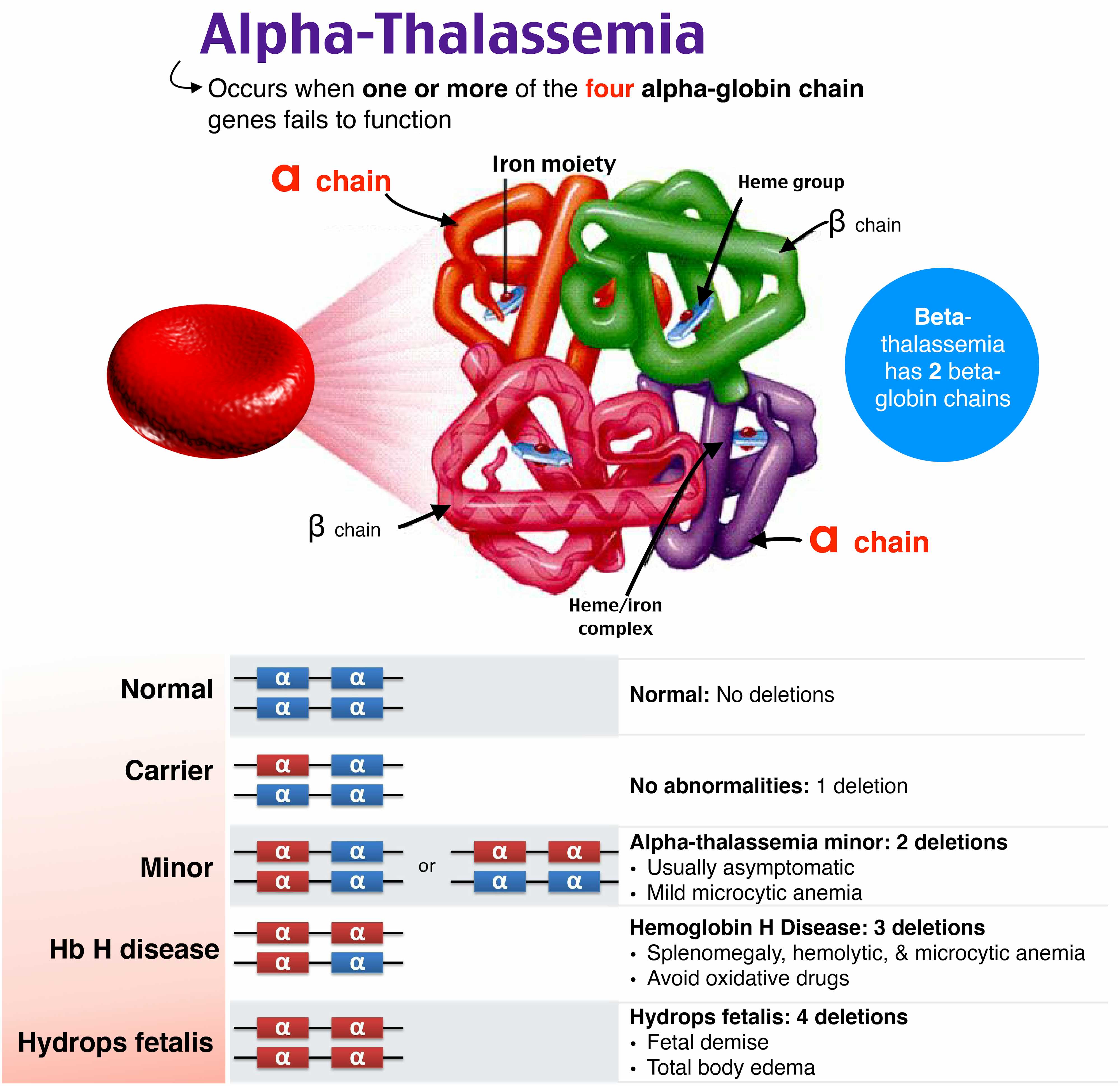
آلفا تالاسمی ناشی از نقص سنتز زنجیره آلفا گلوبین است. وراثت این بیماری به صورت اتوزومال مغلوب است و در نتیجه والدین ناقل به احتمال 25 درصد ممکن است صاحب فرزند بیمار شوند. تشخیص پیش از تولد تالاسمی آلفا در دو مرحله تشخیص ناقلین (مرحله اول) و تشخیص پیش از تولد (مرحله دوم ) انجام می گیرد. در سطح مولکولی، آنها ناشی از حذف جزئی (α+) یا کل (α0) یا به ندرت جهش یک یا چند ژن از چهار ژن آلفاگلوبین هستند. درجدول زیر ژنوتیپ های رایج و طبقه بندی پایه تالاسمی آلفا آورده شده است .
با توجه به تعداد ژنهای تحت تأثیر و از دست دادن عملکرد چهار تصویر بالینی از آلفا تالاسمی وجود دارد:
-
- آلفا تالاسمی حامل(Carrier ) :که از نظر بالینی در فرد قابل تشخیص نیست و در اثر حذف یک زنجیره ی آلفا اتفاق می افتد (α-/αα). این نوع را می توان بر اساس هیپوکرومی خفیف و کاهش قابل اندازه گیری در مقادیر Hb
که در CBC آشکار می شود شناسایی کرد.
-
- تالاسمی مینور: به صورت( - - αα/ یا ( α-/-α که با کم خونی خفیف، هیپوکرومیک میکروسیتیک مشخص می شود .
- بیماری HbH :با سه ژن α غیر فعال، (- -/-α)که کم خونی همولیتیک هیپوکرومیک متوسط همراه با اسپلنومگالی ایجاد می کند و عوارض آن شامل مشکلات قلبی، سنگ کیسه صفرا، زخم ساق پا و کمبود اسید فولیک است.
تصویر فوق نمای هموگلوبین H را در رنگ امیزی حیاتی نشان می دهد.
-
- هموگلوبین بارت :(تالاسمی هموزیگوت)که با کم خونی همولیتیک بسیار جدی و هیدروپس فتالیس جنین بروز میکند و با فقدان هر گونه سنتز زنجیره آلفا-گلوبین (- - /- - ) و کشنده است.
- بتا تالاسمی
تالاسمی بتا در اثر نقص در تولید زنجیره بتا ایجاد میشود. وراثت این بیماری به صورت اتوزومال مغلوب است و در نتیجه والدین ناقل به احتمال 25 درصد ممکن است صاحب فرزند بیمار شوند. تشخیص پیش از تولد تالاسمی بتا در دو مرحله تشخیص ناقلین (مرحله اول) و تشخیص پیش از تولد (مرحله دوم ) انجام می گیرد. افراد نرمال دو نسخه از ژن بتا دارند (β/β) . شدت بیماری بستگی به نوع جهش افراد دارد. بعضی از جهشهای ژن بتا مثل حذفهای ژنی باعث میشود تولید این پروتئین به طور کامل از بین برود (β0) و برخی جهشها مانند جابهجایی هایی که در نواحی تنظیمی ژن اتفاق میافتد منجر به کاهش میزان تولید این پروتئین میشوند (β+).این اختلالات می توانند منجر به خونسازی غیرموثر، کم خونی مزمن، هیپوکسی مزمن و اضافه بار آهن شوند.
با توجه به تعداد ژنهای تحت تأثیر و از دست دادن عملکرد سه تصویر بالینی از بتا تالاسمی وجود دارد که ژنوتیپ های آن های در جدول زیر آورده شده است :
-
- تالاسمی مینور : (بتا تالاسمی هتروزیگوت) که با کم خونی هیپوکرومیک میکروسیتیک خفیف بروز می کند. افراد ناقل (Minor) به سه صورت B0/B, B+/B, B++/B دیده می شوند.
- تالاسمی اینترمدیا : (بتا تالاسمی هتروزیگوت خفیف یا مختلط هتروزیگوت) (B+/B+, B++/B+,B+/B0) با شدت متوسط و با نیاز متفاوت به تزریق خون. عوارض آن عبارتند از بدشکلی های اسکلتی و توده های تومورال در نتیجه اریتروپوزیس عظیم هیپرپلاستیک .
- تالاسمی ماژور: (بتا تالاسمی هتروزیگوت شدید، هموزیگوت یا مختلط) با کم خونی طولانی مدت و وابستگی به انتقال خون بروز می کند..
افراد ماژور β0 /β0) و (β0 /β+ کم خونی شدید دارند . گلبولهای قرمز این افراد ناکارآمد هستند. تولید خون در مغز استخوان های این افراد شدت مییابد که این امر در دراز مدت باعث تغییر شکل استخوانها به ویژه استخوانهای پهن میشود. همچنین این بیماران نیاز به دریافت خون دارند که بیماران را با عوارضی چون افزایش میزان آهن روبهرو میکند. به دلیل کم خونی و بالا رفتن آهن بدن، نارسایی قلبی و آریتمی (ناهماهنگی ضربان قلب) نیز ایجاد می شود. پوست زرد رنگ، تغییرات ناشی از رسوب آهن در کبد، طحال و مثانه دیده میشود.
تشخیص ، انواع اختلات ژنی، یافته های هماتولوژیک و علائم اصلی سندرم های تالاسمی در جدول زیر آورده شده است :

هموگلوبین های غیر طبیعی(ناشی از تغییرات ساختاری)
این گروه از اختلالات هموگلوبین ارثی با توجه به تعویض یک یا چند اسید آمینه در زنجیره(ها)ی پلیپپتیدی α یا β ، تغییراتی کیفی در ساختارشان به نمایش میگذارند؛ این هموگلوبینها را «غیرطبیعی» مینامند یا به صورت دقیقتر «variant hemoglobins». . حدود ۴۰۰ هموگلوبین غیرطبیعی در انسان شناسایی شده است. کم خونی سلول داسی شکل(Hb S)، بیماری هموگلوبین سی(Hb C)، بیماری هموگلوبین اس سی (Hb S + Hb C)، انواعی از این هموگلوبینوپاتی ها هستند که ممکن است عواقب بالینی شدیدی داشته باشند.
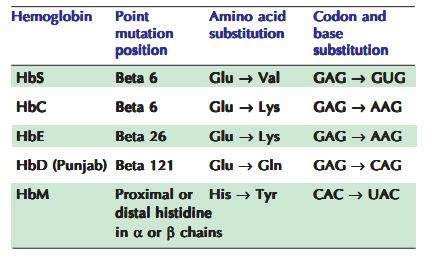
از نظر بالینی اختلالات ساختاری هموگلوبین به چهار گروه زیر تقسیم می شوند:
-
- واریانت های با تمایل به تجمع هموگلوبین و تغییر شکل RBC ها مانند گلبول های قرمز داسی شکل در سندرم داسی .
- واریانت های با سنتز غیر طبیعی هموگلوبین مانند Hb E
- واریانت های با تمایل به رسوب و یا همولیز (هموگلوبین های ناپایدار) مانند Hb Köln
- واریانت های با حمل و نقل غیر طبیعی اکسیژن و پلی سیتمی مادرزادی مانند Hb Johnstown ، یا با سیانوز مادرزادی (مت هموگلوبین های غیر طبیعی و ناهنجاری های HbM).
انواع دیگری از هموگلوبینوپاتیها که ممکن است شناسایی شوند عبارتند از:
- هموگلوبین D
- هموگلوبین G
- هموگلوبین J
- هموگلوبین M
یک فرد می تواند دو ژن غیر طبیعی متفاوت را به ارث ببرد( یکی از هر والدین). این به عنوان هتروزیگوت مرکب یا هتروزیگوت دوگانه شناخته می شود. چندین ترکیب مختلف از نظر بالینی مهم عبارتند از بیماری هموگلوبین SC، بیماری سلول داسی شکل – هموگلوبین دی ، هموگلوبین ای - بتا تالاسمی، و هموگلوبین اس - تالاسمی بتا.
در ادامه به شرح برخی از این هموگلوبینهای غیرطبیعی که اهمیت ویژهی بالینی دارند، پرداخته شده است .
HbS و بیماری سلول داسی شکل
کم خونی داسی شکل اختلال ارثی است که به عنوان بیماری سلول داسی شکل نیز شناخته می شود. گلبول های قرمز معمولاً گرد و انعطاف پذیر هستند، بنابراین به راحتی در رگ های خونی حرکت می کنند. در کم خونی داسی شکل، برخی از گلبول های قرمز به شکل داسی یا هلال ماه به وجود می آیند. این سلول های داسی شکل سفت و چسبناک می شوند که می تواند جریان خون را کند یا مسدود کند. این سلول ها به اندازه گلبول های قرمز معمولی، گرد و قرمز دوام نمی آورند که منجر به کم خونی (تعداد کم گلبول های قرمز خون) می شود.
علائم و نشانه های بیماری سلول داسی شکل معمولا در اوایل دوران کودکی شروع می شود و ممکن است شامل کم خونی، عفونت های مکرر و دوره های دوره ای درد (به نام بحران) باشد. شدت علائم از فردی به فرد دیگر متفاوت است. برخی از افراد علائم خفیف دارند، در حالی که برخی دیگر اغلب به دلیل عوارض جدی تر در بیمارستان بستری می شوند. این بیماری در اثر تغییرات ژنتیکی در ژن HBB ایجاد می شود و با الگوی اتوزومی مغلوب به ارث می رسد. یکی از عوارض جدی بیماری سلول داسی، فشار خون بالا در رگ های خونی تامین کننده ریه ها (فشار خون ریوی) است که می تواند منجر به نارسایی قلبی شود. فشار خون ریوی در حدود ۱۰ درصد از بزرگسالان مبتلا به بیماری سلول داسی شکل رخ می دهد. بیمارانی که تحت درمان بهینه قرار می گیرند می توانند طول عمری بین 50 تا 60 سال داشته باشند.
ناقل های ژن HbS هتروزیگوت از نظر بالینی یا خونی تحت تأثیر قرار نمی گیرند
با توجه به نامگذاری بین المللی، اصطلاح رایج قبلی "کم خونی داسی شکل" نباید استفاده شود، زیرا جنبه های غالب این بیماری، انسداد عروق و آسیب اندام ناشی از آن است، نه کم خونی.

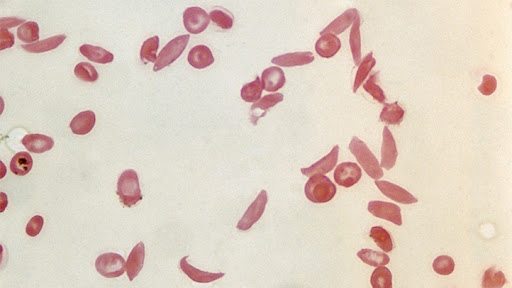
شایعترین انواع بیماری سلول های داسی شکل
بیماری سلول های داسی شکل در اثر به ارث بردن ژن معیوب از والدین ایجاد میشود. این بیماری بر اساس اینکه فرزند چه تعداد ژن معیوبی را از والدین خود به ارث برده است، دارای انواع مختلفی است که در ادامه به آنها اشاره شده است.
- بیماری سلول های داسی شکل، نوعِ SS (بیماری هموگلوبین SS)
بیماری هموگلوبین SS رایجترین و شدیدترین نوع بیماری سلول داسی شکل است. زمانی که هم پدر و هم مادر ناقل ژن معیوب هموگلوبین S باشند و آنرا به فرزند خود انتقال دهند، شخص به بیماری سلول های داسی شکل نوعِ SS مبتلا خواهد بود. این بیماری کم خونی (آنمی) داسی شکل[3] نیز نامیده میشود.
- صفت سلول های داسی شکل
چنانچه فردی یک ژن معیوب بیماری سلول های داسی شکل را از یکی از والدین خود به ارث برده و از دیگری یک ژن سالم را به ارث ببرد، به صفت سلول های داسی شکل مبتلا خواهد بود. به این معنا که فرد اغلب هیچ علائمی از بیماری را ندارد اما قادر به انتقال این ژن معیوب به فرزندان خود است.
- بتا تالاسمی داسی شکل SB
این بیماری به این علت در افراد ایجاد میشود که فرد از والدین خود یک ژن بتاتالاسمی و یک ژن سلول داسی شکل به ارث ببرد. شدت بیماری با توجه به میزان تولید بتا گلوبین طبیعی متفاوت است. هنگامی که هیچ بتاگلوبینی در بدن فرد تولید نشود، علائم بیماری تقریباً مشابه کم خونی سلول داسی شکل خواهد بود.
- بیماری داسی شکل ترکیبی هموگلوبینSC، هموگلوبین SD، هموگلوبین SE و هموگلوبین SO
همانطور که گفته شد، گونههای غیرطبیعی دیگری از هموگلوبین نیز ممکن است به دلیل جهش ژنتیکی در بدن افراد ایجاد شود. زمانی که فردی هریک از این ژنهای معیوب را به همراه یک ژن سلول داسی شکل از والدین خود به ارث ببرد، به بیماری هموگلوبینSC، هموگلوبین SD، هموگلوبین SE و هموگلوبین SO مبتلا خواهد بود. البته این چند مورد، در بین انواع دیگر کمتر رایج بوده و به ندرت اتفاق میافتد.
ناهنجاری HbC و بیماری HbC
بیماری هموگلوبین C (که در افرادی که دو نسخه از ژن دارند دیده می شود) نادر و نسبتاً خفیف است. معمولاً باعث کم خونی همولیتیک خفیف و بزرگ شدن خفیف تا متوسط طحال می شود. هموزیگوسیتی HbC، یا بیماری HbC، به روشی مشابه بیماری سلول داسی شکل پیشرفت می کند، اما جدی تر است . کم خونی همولیتیک متغیر غالب ترین شکل این بیماری است. حاملان ژن HbC هتروزیگوت از سلامت بالینی کامل برخوردار هستند .
بیمار مبتلا به بیماری ترکیبی هموگلوبین C و سلول داسی (HbSC) می تواند به رتینوپاتی عروقی، نکروز آواسکولار و ترومبوز میکروواسکولار مدولاری کلیه مبتلا شود..
ناهنجاری HbE و بیماری HbE
این یکی از رایج ترین انواع هموگلوبین زنجیره بتا در جهان است. در افراد آسیای جنوب شرقی بسیار شایع است. افرادی که از نظر Hb E هموزیگوت هستند، عموماً دارای کم خونی همولیتیک خفیف، گلبول های قرمز میکروسیتیک و بزرگ شدن خفیف طحال هستند. یک کپی از ژن هموگلوبین E علائمی ایجاد نمی کند مگر اینکه با جهش دیگری (مثلاً جهشی که باعث بتا تالاسمی می شود) ترکیب شود.
Hb E هموگلوبینی ناپایدار است و اغلب با تالاسمیها ترکیب میشود که ممکن است منجر به هموگلوبینوپاتیهای جدی شود .
تشخیص ، انواع اختلات ژنی، یافته های هماتولوژیک و علائم اصلی واریانت های هموگلوبین در جدول زیر آورده شده است .
شده است .
تشخیص و آزمایش بررسی هموگلوبینوپاتی ها :
زمانی آزمایش هموگلوبینوپاتی درخواست می شود که پزشک مشکوک باشد که علائم و نشانههای فرد در نتیجه تولید غیرطبیعی هموگلوبین است. اشکال هموگلوبینوپاتی اغلب منجر به کم خونی همولیتیک می شود که منجر به علائم و نشانه هایی مانند:
ضعف، خستگی ،کمبود انرژی،زردی ،پوست رنگ پریده
آزمایش ممکن است برای موارد زیر استفاده شود:
غربالگری: همه نوزادان برای انواع خاصی از هموگلوبینوپاتی غربالگری می شوند. غربالگری قبل از تولد اغلب بر روی والدین پرخطر با پیشینه قومی مرتبط با شیوع بالاتر ناهنجاری هموگلوبین و آنهایی که اعضای خانواده آسیب دیده دارند انجام می شود. غربالگری همچنین ممکن است همراه با مشاوره ژنتیک قبل از بارداری انجام شود تا مشخص شود که آیا والدین ناقل هستند یا خیر.
تشخیص: برای تشخیص و/یا شناسایی هموگلوبینوپاتی (ناهنجاری هموگلوبین یا تالاسمی) در افرادی که علائم کم خونی غیرقابل توضیح یا نتایج غیرطبیعی در شمارش کامل خون (CBC) دارند.
آزمایش بررسی هموگلوبینوپاتی ها به ویژه در شرایط زیر باید انجام شود :
-
- کم خونی هیپوکرومیک میکروسیتیک که پس از کمبود آهن رد شده است
- کم خونی همولیتیک مزمن
- بحران انسداد عروقی با علت نامشخص در بیماران مناطقی که HbS و/یا HbC در آنها گسترده است.
- کم خونی ناشی از دارو
- اریتروسیتوز و/یا سیانوز ناشی از عوامل خونی
- هیدروپس جنینی با علت نامشخص
- پیشگیری (آزمایش اعضای خانواده، تشخیص همسر برای مشاوره ژنتیک)
- تشخیص قبل از تولد
برخی از اشکال شدید هموگلوبینوپاتی ها (مانند بیماری سلول داسی شکل) ممکن است منجر به علائم و نشانه های جدی مانند دوره های درد شدید، تنگی نفس، بزرگ شدن طحال و مشکلات رشد در کودکان شود.
ارزیابی هموگلوبینوپاتی برای تشخیص اشکال غیرطبیعی و/یا مقادیر نسبی هموگلوبین، ، استفاده میشود.
چندین روش آزمایشگاهی مختلف برای ارزیابی هموگلوبینوپاتی موجود در فرد وجود دارد. برخی از این موارد عبارتند از:
الکتروفورز هموگلوبین (Hb Electrophoresis)
تمرکز ایزوالکتریک هموگلوبین (Hb IsoElectricFocusing )
هموگلوبین توسط کروماتوگرافی مایع با کارایی بالا (HPLC)
هموگلوبین توسط الکتروفورز ناحیه مویرگی (CapillaryZoneElectrophoresis)
هموگلوبین توسط طیف سنجی جرمی
تست حلالیت هموگلوبین(solubility test): به طور خاص برای آزمایش هموگلوبین S )هموگلوبین اصلی در بیماری سلول داسی شکل (استفاده می شود.
این روش ها انواع مختلف هموگلوبینوپاتی را بر اساس خواص فیزیکی و شیمیایی مولکول های مختلف هموگلوبین ارزیابی می کنند. بسیاری از انواع رایج هموگلوبین یا تالاسمی ها را می توان با استفاده از یکی از این آزمایش ها یا ترکیبی از آنها شناسایی کرد. مقادیر نسبی هر نوع هموگلوبین شناسایی شده می تواند به تشخیص کمک کند. با این حال، یک آزمایش منفرد معمولاً برای تشخیص هموگلوبینوپاتی کافی نیست. بلکه نتایج چندین آزمایش مختلف در نظر گرفته می شود. نمونه هایی از سایر آزمایش ها که ممکن است انجام شوند عبارتند از:
CBC
لام خون محیطی( PBS)
رنگ آمیزی فوق حیاتی (Supravital stain )
مطالعات آهن مانند آهن سرم، TIBC، ترانسفرین، فریتین
آزمایش ژنتیکی و DNA
- شمارش کامل خون CBC
کلید تشخیص و شناسایی موفقیت آمیز هموگلوبینوپاتی ها، به ویژه تالاسمی ها، داده های اولیه هماتولوژیک است. سرنخ تالاسمی با MCV پایین (میانگین حجم RBC) یا MCH )متوسط هموگلوبین گلبولی) پایین یا هر دو همراه است. اگرچه کمبود آهن توضیح دیگری برای MCV یا MCH پایین است و جهت افتراق باید بررسی شود. بنابراین اولین قدم بعد از شمارش غیرطبیعی اولیه خون، رد کردن تشخیص کمبود آهن و در صورت وجود، درمان آن است. سپس شمارش خون تکرار می شود و اگر MCV/MCH پایین بماند، احتمال تالاسمی وجود دارد نیازمند بررسی است .
لازم به ذکر است که در بعضی شرایط، سرنخ اصلی تالاسمی ممکن است پنهان شود زیرا MCV به طور کاذب طبیعی یا حتی افزایش یافته است به عنوان مثال MCV در شرایطی مانند کمبود ویتامین B12 و اسید فولیک و همچنین در بیماران مبتلا به HIV که آنالوگهای نوکلئوزیدی مصرف میکنند بالا می رود بنابر این همزمانی تالاسمی با موارد فوق ممکن است میزان این پارامترMCV نرمال یا حتی بالا باشد
یک هموگلوبینوپاتی مهم وجود دارد که اگر فقط از MCV یا MCH برای غربالگری اولیه استفاده شود، نادیده گرفته می شود. این ناقل HbS است. بنابراین، متخصصان بهداشت یا آزمایشگاههایی که با جمعیتهایی که HbS در آنها رخ میدهد سر و کار دارند، همیشه باید الکتروفورز هموگلوبین را با درخواست شمارش کامل خون در نظر بگیرند.
- اسمیر خون محیطی :
به عنوان بخشی از روتین شمارش کامل خون، یک اسمیر محیطی خون می تواند مفید باشد زیرا ممکن است سرنخی از وجود بیماری سلول داسی شکل (HbS) یا Hb ناپایدار ارائه دهد. تغییراتی مانند سلول های هدف (Target cell) و بازوفیلیک استیپلینگ در اسمیر دیده شود که البته ممکن است به طور قطعی با هموگلوبینوپاتی مرتبط نباشد ، اما اگر در این موارد ، MCV یا MCH نیز پایین بود، یافته های مفیدی به نفع تالاسمی خواهند بود.
برخی MCH را نسبت به MCV به عنوان پارامتر کلیدی تر گلبول قرمز ترجیح می دهند زیرا در صورت تاخیر در پردازش خون برای آزمایش، MCV می تواند تغییر کند.
RDW ( دامنه پراکندگی گلبول قرمز از نظر اندازه ) اندازه گیری می کند. در فقر آهن بیشتر است اما در تالاسمی ها خیر و بنابراین می تواند در تشخیص اینکه کدام احتمال بیشتر است کمک کند. با این حال، به ویژه در دوران بارداری، یافتن هر دو غیرعادی نیست.
برخی از هموگلوبینوپاتی ها، به ویژه HbS، دارای MCV طبیعی و MCH طبیعی خواهند بود و بنابراین اگر از شمارش کامل خون به عنوان یک آزمایش غربالگری استفاده شود، از دست خواهند رفت.
از آنجایی که تعویض بتا گلوبین جنین به بزرگسال معمولاً تا حدود 6 ماهگی کامل نیست، تشخیص بتا تالاسمی در نوزاد بر اساس شمارش کامل خون دشوار است. با این حال، یک هماتولوژیست اطفال با تجربه در بررسی فیلم های خونی و ارزیابی تالاسمی ممکن است بر اساس پارامترهای خونی و تغییرات گلبول قرمز در فیلم خون حدس منطقی بزند که آیا این تغییرات نشان دهنده تالاسمی هستند یا خیر.
در حاملگی های پرخطر که با تشخیص قبل از تولد نظارت نمی شود (تشخیص دیرهنگام یا عدم علاقه زوجین به آزمایشات قبل از تولد)، نگهداری مقداری از خون بند ناف برای انجام آزمایش DNA مهم است.
- مطالعات آهن
اگرچه پیشنهاد شده است که کمبود آهن در برخی جوامع با خطر ابتلا به تالاسمی غیرمعمول است، اما ضروری است که کمبود آهن همیشه قبل از انجام هر گونه آزمایش بیشتر در کارهای هموگلوبینوپاتی کنار گذاشته شود. بنابراین، سطوح فریتین (و در صورت لزوم آهن سرم، ظرفیت اتصال آهن و درصد اشباع) جستجو می شود. به دلیل اینکه در برخی مواقع، به ویژه در دوران بارداری، ممکن است ذخایر آهن کم شود و یا در صورت کمبود آهن، MCV یا MCH تحت تأثیر کمبود آهن باشد بررسی پروفایل آهن اهمیت دارد . همچنین گاهی دیده می شود که سطح HbA2 می تواند به طور کاذب در اثر کمبود آهن کاهش یابد. در صورت وجود کمبود آهن، اصلاح آن ضروری است و سپس شمارش کامل خون و سایر بررسی ها تکرار می شود.
- آزمایشات DNA
ممکن است برای تشخیص جهش در ژنهایی که زنجیرههای پروتئینی که هموگلوبین را تشکیل میدهند، انجام شود این یک آزمایش روتین نیست، اما می تواند برای تأیید اینکه آیا یک فرد دارای ژن جهش یافته است و آیا یک یا دو نسخه جهش یافته (هتروزیگوت یا هموزیگوت) وجود دارد یا خیر، استفاده شود.
این آزمایشات در دو حالت درخواست می شود:
(1) زمانی که هموگلوبینوپاتی را نمی توان با آزمایش های خونی خاص تأیید کرد( گاهی اوقات می توان به هموگلوبینوپاتی مشکوک شد اما آزمایشات هماتولوژی و همچنین مطالعات خانوادگی نمی توانند تعیین کنند که کدام ژن احتمالاً درگیر است).
(2) برای جستجوی جهش زمینه ای در هموگلوبینوپاتی که معمولاً به عنوان بخشی از یک بررسی قبل از تولد، مورد نیاز است.
به طور کلی، تالاسمیα در اثر حذف ژن ایجاد می شود، اگرچه به طور فزاینده ای اشکال غیر حذفی تالاسمی α در موارد دشوار جستجو می شود. در مقابل، بتا تالاسمی ها و واریانت های هموگلوبین در بیشتر موارد، نتیجه جهش های نقطه ای هستند. بنابراین، استراتژیهای بررسی جهش DNA با آگاهی از نقص احتمالی زمینهای که نیاز به شناسایی دارد، حائز اهمیت است.
پیشنهاد ما به شما عزیزان
همانطور که اشاره کردیم، آزمایش الکتروفورز هموگلوبین منجر به تشخیص هرگونه هموگلوبین غیرعادی، نشات گرفته از اختلالات ژنتیکی می شود. علاوه بر این، نوزادان نیز به صورت پیوسته از نظر وجود اختلالات ژنتیکی- هموگلوبینی تحت غربالگری و نظارت قرار می گیرند. اگر سابقه هموگلوبین غیرعادی یا کم خونی به هر دلیلی در شما وجود داشته باشد، پزشک درخواست آزمایش الکتروفورز هموگلوبین و سایر تست های تکمیلی و ژنتیک را ابراز می کند.
همچنین اگر شما قصد بچه دار شدن دارید، برای بررسی وجود هموگلوبین غیر طبیعی در خون خود باید مشاور ژنتیک انجام دهید.
این کار به شما کمک می کند تا متوجه شوید که فرزند شما مبتلا به نوع خاصی از کم خونی ارثی خواهد بود یا خیر.
آزمایشگاه تشخیص طبی راز با در اختیار داشتن چندین دستگاه Sebia Capilarys Electrophoresis قادر به ارائه خدمات ارزشمندی در زمینه تستهای تشخیصی با متد الکتروفورز با دقت بالا و در بازه زمانی مناسب، میباشد. معتبرترین سازنده تجهیزات تمام اتوماتیک الکتروفورز سازنده سیستمهای Clinical Capilary Electrophoresis در جهان، Sebia Electrophoresis میباشد که پیشرفتهترین سیستم جداسازی هموگلوبینها را با توانایی تفکیک بیش از ۱۵۰ واریانت مختلف دارد. همچنین C.Z.E (Cpillary Zone Electrophoresis) دارای تاییدیه CE و FDA میباشد.
آزمایشگاه راز یکی از بزرگترین و مجهزترین آزمایشگاه های پزشکی در شهر شیراز می باشد و انواع آزمایشات پاتولوژی ، تشخیص طبی و مشاوره ژنتیک را نیز انجام می دهد.
این آزمایشگاه دارای امکانات، تجهیزات مدرن و فضای آزمایشگاهی وسیع تری بوده و در این آزمایشگاه در هر بخش، افراد متخصص مشغول انجام تست های مختلف هستند.
تهیه کننده :
مرضیه آزاد ، کارشناس ارشد ایمنی شناسی پزشکی -واحد تحقیق و توسعه آزمایشگاه پاتوبیولوژی راز
زیر نظر :
دکتر عزت اله اسدی ، متخصص پاتولوژی بالینی- آزمایشگاه راز
رفرنس :
https://labpedia.net/anemia-part-5-a-sickle-cell-anemia-discussion-and-workup/
Hemoglobinopathies: clinical manifestations, diagnosis, and treatment Elisabeth Kohne 1 Affiliations expand PMID: 21886666 DOI: 10.3238/arztebl.2011.0532
https://go.roshreview.com/obgyn-qid-166132
Hemoglobin C Disease Bibek Karna; Suman K. Jha; Eiman Al Zaabi.Author Information and Affiliations Last Update: May 29, 2023.