


Hemoglobinopathy
Hemoglobinopathies are genetic disorders that affect the structure or production of hemoglobin.
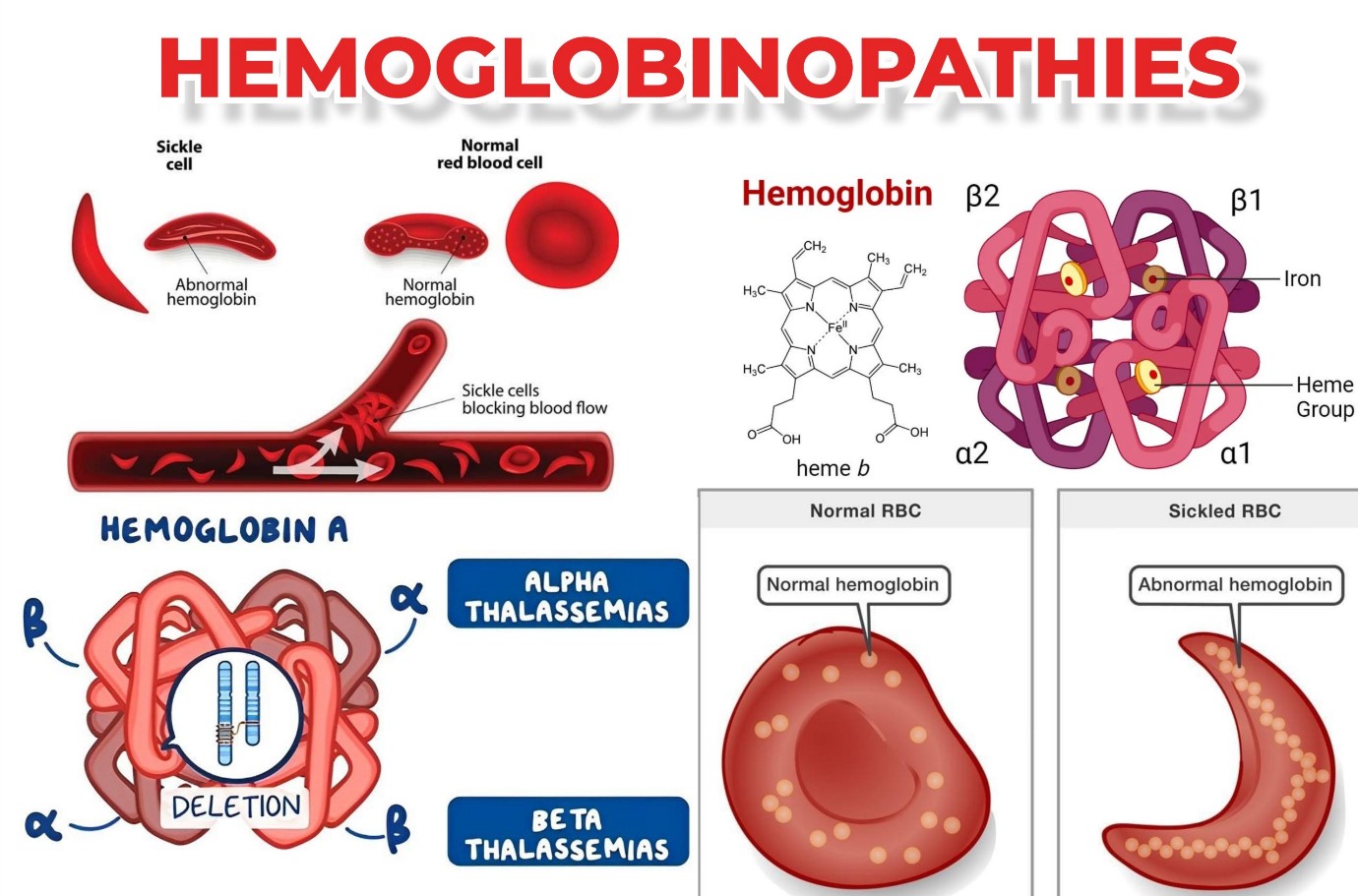
Hemoglobin (Hb) is an iron-containing protein found in all red blood cells (RBCs) and binds to the oxygen molecule in the lungs, allowing the red blood cells to deliver oxygen to all cells and tissues in the body. Hemoglobin consists of a part called heme, a molecule with central iron, and another part consisting of four globin (protein) chains. Globin chains have different names depending on their structure: alpha, beta, gamma and delta. The type of globin chains play an essential role in the function of hemoglobin and its ability to transport oxygen.
The natural types of hemoglobin are:
Hemoglobin A: It makes up about 95-98% of the hemoglobin in adults, which includes two alpha chains and two beta chains.
Hemoglobin: A2 makes up about 2-3% of hemoglobin in adults. It has two alpha protein chains and two delta chains.
Hemoglobin F Fetal hemoglobin: 1 to 2% of hemoglobin in adults. It has two alpha chains and two gamma chains. This hemoglobin is the first hemoglobin produced during pregnancy. Its production usually decreases shortly after birth and reaches adult levels at 1-2 years of age.
The term "hemoglobinopathy" includes all genetic disorders of hemoglobin, which are divided into two main groups as follows, both of which are caused by mutations or deletions or a combination of the two in the α or β globin gene:
Thalassemia syndromes: When a gene defect causes disorders of Hb synthesis, this leads to thalassemia. The structure of hemoglobin is normal in these cases.
Hemoglobin structural disorders (abnormal hemoglobins). It is when changes in the structure of Hb lead to the creation of abnormal hemoglobin.
There are also many hybrid forms that combine features of both groups, for example alpha thalassemia-hemoglobin E combination.
Thalassemia syndromes
It includes disorders of Hb synthesis. Thalassemia syndromes are caused by the insufficient production of 1 or 2 types of globin chains and with its type (alpha, beta, delta, gamma) and the amount of insufficient hemoglobin production (number of defective genes) and the severity of clinical symptoms (partial , intermediate, major) is identified. .
Types of thalassemia syndromes:
Alpha thalassemia
Alpha thalassemia is caused by a defect in the synthesis of the alpha globin chain. The inheritance of this disease is autosomal recessive, and as a result, carrier parents have a 25% chance of having a sick child. Prenatal diagnosis of alpha thalassemia is carried out in two stages: carrier diagnosis (first stage) and prenatal diagnosis (second stage). At the molecular level, they result from a partial (α+) or total (α0) deletion or, rarely, a mutation of one or more of the four alphaglobin genes. The table below shows the common genotypes and basic classification of alpha thalassemia.
According to the number of affected genes and the loss of function, there are four clinical pictures of alpha thalassemia:
Alpha thalassemia carrier (Carrier): which is clinically undetectable in a person and occurs due to the deletion of an alpha chain (α-/αα). This type can be diagnosed based on mild hypochromia and a measurable decrease in Hb values
identified in CBC.
Thalassemia minor: in the form of (--αα/ or (α-/-α), which is characterized by mild, hypochromic, microcytic anemia.
HbH disease: with three inactivated α genes, (- -/-α) which causes moderate hypochromic hemolytic anemia with splenomegaly, and its complications include heart problems, gallstones, leg ulcers, and folic acid deficiency. .
The image above shows the appearance of hemoglobin H in vital staining.
Hemoglobin Barrett (homozygous thalassemia) which occurs with very serious hemolytic anemia and hydrops fetalis of the fetus and with the absence of any alpha-globin chain synthesis (- - /-) and is fatal.
Beta thalassemia
Beta thalassemia is caused by a defect in the production of the beta chain. The inheritance of this disease is autosomal recessive, and as a result, carrier parents have a 25% chance of having a sick child. Prenatal diagnosis of beta thalassemia is carried out in two stages: carrier diagnosis (first stage) and prenatal diagnosis (second stage). Normal people have two copies of the beta gene (β/β). The severity of the disease depends on the type of mutation. Some mutations of the beta gene, such as gene deletions, cause the production of this protein to be completely lost (β0), and some mutations, such as translocations that occur in the regulatory regions of the gene, lead to a decrease in the production of this protein (β+). These disorders They can lead to ineffective hematopoiesis, chronic anemia, chronic hypoxia and iron overload.