


اعتلال الهيموجلوبين
اعتلالات الهيموجلوبين هي اضطرابات وراثية تؤثر على بنية الهيموجلوبين أو إنتاجه.
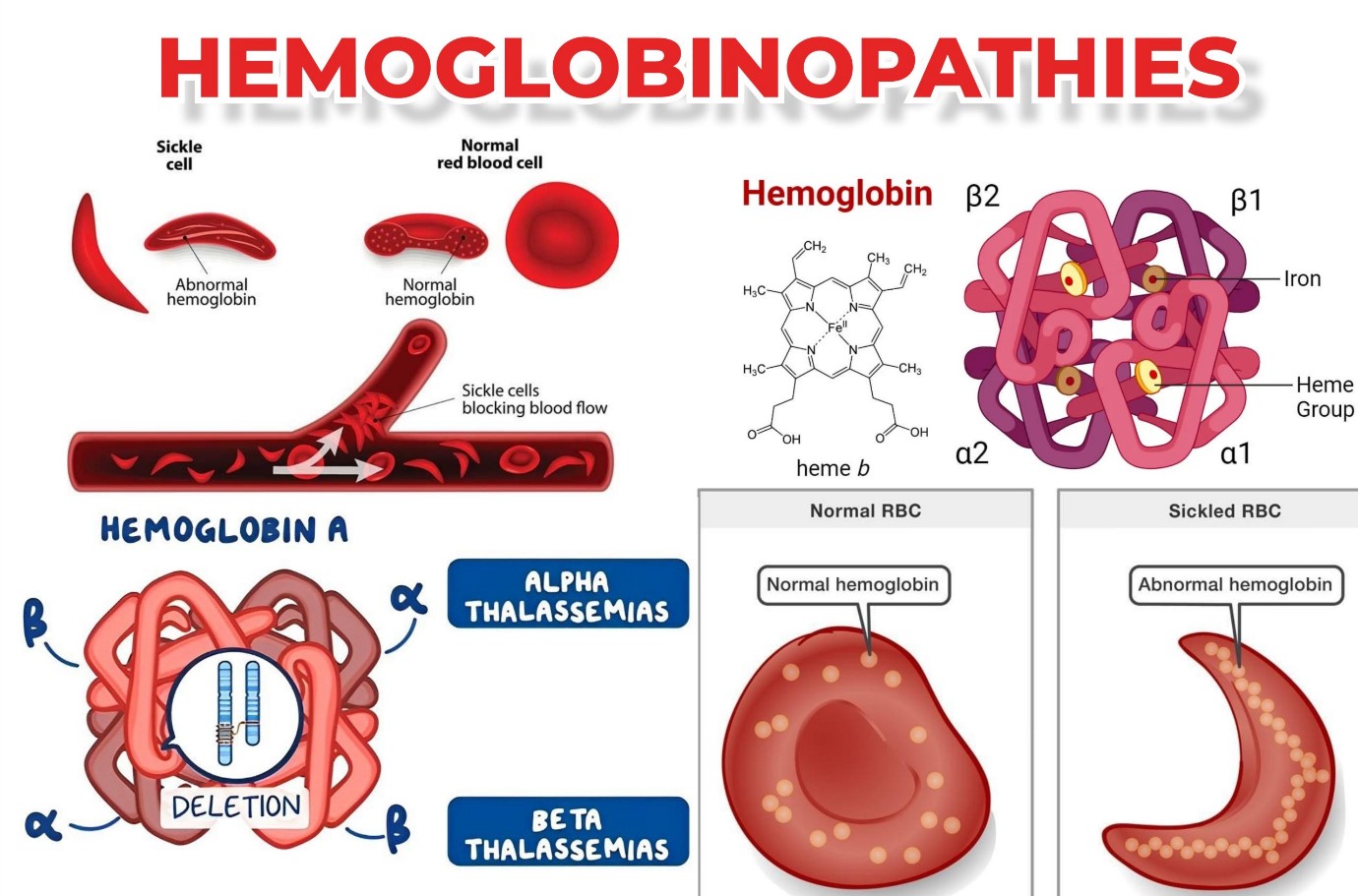
الهيموجلوبين (Hb) هو بروتين يحتوي على الحديد ويوجد في جميع خلايا الدم الحمراء (RBCs) ويرتبط بجزيء الأكسجين في الرئتين، مما يسمح لخلايا الدم الحمراء بتوصيل الأكسجين إلى جميع الخلايا والأنسجة في الجسم. يتكون الهيموجلوبين من جزء يسمى الهيم، وهو جزيء به حديد مركزي، وجزء آخر يتكون من أربع سلاسل الجلوبين (البروتين). سلاسل جلوبين لها أسماء مختلفة اعتمادا على بنيتها: ألفا وبيتا وغاما ودلتا. ويلعب نوع سلاسل الجلوبين دورا أساسيا في وظيفة الهيموجلوبين وقدرته على نقل الأكسجين.
الأنواع الطبيعية للهيموجلوبين هي:
الهيموجلوبين أ: يشكل حوالي 95-98% من الهيموجلوبين عند البالغين، وهو يشتمل على سلسلتي ألفا وسلسلتي بيتا.
الهيموجلوبين: يشكل A2 حوالي 2-3% من الهيموجلوبين لدى البالغين. يحتوي على سلسلتين من بروتين ألفا وسلسلتين دلتا.
الهيموجلوبين F: الهيموجلوبين الجنيني: 1 إلى 2% من الهيموجلوبين عند البالغين. لديها سلسلتين ألفا وسلسلتين جاما. هذا الهيموجلوبين هو الهيموجلوبين الأول الذي يتم إنتاجه أثناء الحمل. يتناقص إنتاجه عادة بعد فترة قصيرة من الولادة ويصل إلى مستويات البالغين عند عمر 1-2 سنة.
يشمل مصطلح "اعتلال الهيموجلوبين" جميع الاضطرابات الوراثية للهيموجلوبين، والتي تنقسم إلى مجموعتين رئيسيتين على النحو التالي، وكلاهما ناتج عن طفرات أو عمليات حذف أو مزيج من الاثنين في جين α أو β جلوبين:
متلازمات الثلاسيميا: عندما يسبب خلل في الجينات اضطرابات في تخليق الهيموجلوبين، يؤدي ذلك إلى الإصابة بالثلاسيميا. تكون بنية الهيموجلوبين طبيعية في هذه الحالات.
الاضطرابات الهيكلية للهيموجلوبين (الهيموجلوبين غير الطبيعي). يحدث ذلك عندما تؤدي التغييرات في بنية Hb إلى تكوين الهيموجلوبين غير الطبيعي.
كما أن هناك العديد من الأشكال الهجينة التي تجمع بين سمات المجموعتين، على سبيل المثال تركيبة ألفا ثلاسيميا والهيموجلوبين E.
متلازمات الثلاسيميا
وتشمل اضطرابات تخليق الهيموجلوبين (Hb) التي تنتج عن عدم كفاية إنتاج نوع أو نوعين من سلاسل الجلوبين ونوعه (ألفا، بيتا، دلتا، جاما) وكمية إنتاج الهيموجلوبين غير الكافي (عدد الجينات المعيبة) و يتم تحديد شدة الأعراض السريرية (الجزئية والمتوسطة والكبيرة). .
أنواع متلازمات الثلاسيميا:
ألفا ثلاسيميا
يحدث مرض ثلاسيميا ألفا بسبب خلل في تركيب سلسلة ألفا جلوبين. وراثة هذا المرض هي صبغية جسمية متنحية، ونتيجة لذلك، فإن الوالدين الحاملين للمرض لديهم فرصة بنسبة 25٪ لإنجاب طفل مريض. يتم التشخيص قبل الولادة لمرض ألفا ثلاسيميا على مرحلتين: تشخيص حامل المرض (المرحلة الأولى) والتشخيص قبل الولادة (المرحلة الثانية). على المستوى الجزيئي، تنتج عن حذف جزئي (α+) أو كلي (α0) أو، نادرًا، طفرة في واحد أو أكثر من جينات ألفاجلوبين الأربعة. يوضح الجدول أدناه الأنماط الجينية الشائعة والتصنيف الأساسي لمرض ألفا ثلاسيميا.
وفقًا لعدد الجينات المصابة وفقدان الوظيفة، هناك أربع صور سريرية لمرض ألفا ثلاسيميا:
حامل الثلاسيميا ألفا (Carrier): وهو غير قابل للاكتشاف سريريا لدى الشخص ويحدث بسبب حذف سلسلة ألفا (α-/αα). يمكن تشخيص هذا النوع بناءً على نقص صبغة الدم الخفيف وانخفاض قابل للقياس في قيم Hb
تم تحديدها في CBC.
الثلاسيميا الصغرى: على شكل (--αα/ أو (α-/-α)، ويتميز بفقر دم خفيف ناقص الصباغ صغير الكريات.
مرض HbH: مع ثلاثة جينات α معطلة، (- -/- α) الذي يسبب فقر الدم الانحلالي ناقص الصباغ المعتدل مع تضخم الطحال، وتشمل مضاعفاته مشاكل في القلب، وحصوات المرارة، وتقرحات الساق، ونقص حمض الفوليك.
الصورة أعلاه توضح ظهور الهيموجلوبين H في الصبغة الحيوية.
الهيموجلوبين باريت (الثلاسيميا المتماثلة اللواقح) الذي يحدث مع فقر الدم الانحلالي الخطير للغاية واستسقاء الجنين ومع عدم وجود أي تخليق لسلسلة ألفا جلوبين (- - /-) وهو مميت.
بيتا ثلاسيميا
بيتا ثلاسيميا سببه خلل في إنتاج سلسلة بيتا. وراثة هذا المرض هي صبغية جسمية متنحية، ونتيجة لذلك، فإن الوالدين الحاملين للمرض لديهم فرصة بنسبة 25٪ لإنجاب طفل مريض. يتم إجراء التشخيص قبل الولادة لمرض بيتا ثلاسيميا على مرحلتين: تشخيص الحامل (المرحلة الأولى) والتشخيص قبل الولادة (المرحلة الثانية). لدى الأشخاص الطبيعيين نسختان من جين بيتا (β/β). تعتمد شدة المرض على نوع الطفرة. بعض طفرات جين بيتا، مثل عمليات حذف الجينات، تتسبب في فقدان إنتاج هذا البروتين بشكل كامل (β0)، كما تؤدي بعض الطفرات، مثل عمليات النقل التي تحدث في المناطق التنظيمية للجين، إلى انخفاض إنتاج هذا البروتين. من هذا البروتين (β+). هذه الاضطرابات يمكن أن تؤدي إلى تكون الدم غير فعالة، وفقر الدم المزمن، ونقص الأكسجة المزمن والحديد الزائد.